Aortopulmonary Window (AP Window)
Gary Dhillon, MD
Overview and Natural History
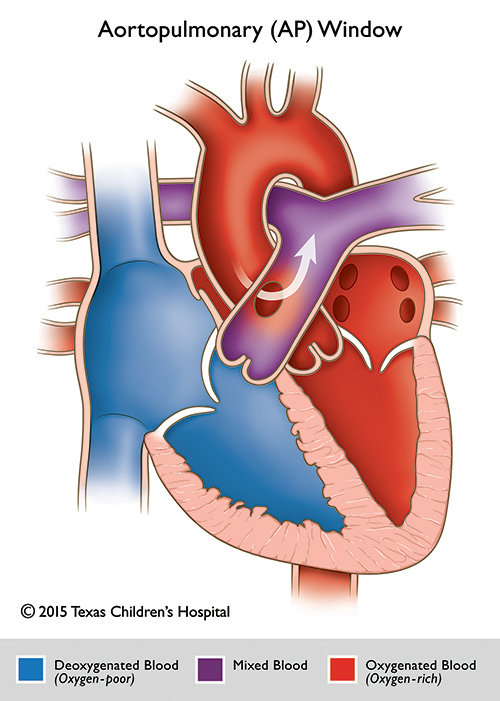
Aortopulmonary window (AP window) is a congenital heart defect characterized by a communication between the ascending aorta and the pulmonary artery in the presence of separate semilunar valves, an right ventricular outflow tract, and separate arterial trunks.
Epidemiology
It is a rare malformation present in less than 0.5% of the congenital heart disease population. AP window is associated with other cardiac defects in 25-50% of cases. These associated anomalies include: Tetralogy of Fallot, aortic arch anomalies (coarctation, type A interrupted aortic arch), anomalous origin of the coronary arteries, tricuspid atresia, aortic or pulmonary atresia, and transposition of the great arteries.
Embryology and Genetics:
The aortopulmonary septum develops as a wedge of tissue (capped by neural crest cells) that grows ventrally from the dorsal wall of the aortic sac between the origins of the fourth and sixth aortic arches. This structure fuses with the distal margins of the embryonic outflow cushions (also capped by neural crest cells) to close the embryonic aortopulmonary foramen. Failure to close this foramen by fusion of the neural crest cells results in an aortopulmonary window.
Unlike the other conotruncal anomalies, aortopulmonary window has not been reported in association with DiGeorge syndrome (22q11 microdeletion syndrome), other velocardiofacial syndromes, and the risk of associated chromosomal anomalies in aortopulmonary window appears low. There are no known genetic or environmental risk factors associated with aortopulmonary window documented to date.
Hemodynamics
The physiology of an AP window is a left-to-right shunting lesion; therefore, when this malformation is isolated and large, patients may present with: pulmonary overcirculation, congestive heart failure, and development of pulmonary vascular disease with pulmonary hypertension. Although AP windows typically lead to pulmonary overcirculation in a normally-saturated patient, cyanosis can be seen with large defects due to bidirectional shunting and mixing at the arterial level. Cyanosis may also be seen with AP window if associated with a cyanotic congenital heart disease lesions earlier in neonatal life when pulmonary vascular resistance is still high and mixing at the arterial level is not yet occurring. Due to the extent of pulmonary overcirculation and exposure of the pulmonary vasculature to systemic arterial pressures, earlier surgical repair is recommended to prevent irreversible pulmonary vascular disease.
Types
AP window defects are separated into different subtypes based on location and arterial structures involved:
Type I: This defect is more proximally located between the origin of the main pulmonary artery and the ascending aorta immediately above the sinus of Valsalva (due to deficient septation of the aortopulmonary trunk during development) with little inferior rim separating the AP window from the semilunar valves. These defects tend to be large, round or oval shaped, and are more often seen.
Type II: This defect is more distal, between the ascending aorta and the origin of the right pulmonary artery (due to abnormal migration of the 6th aortic arch during development). These defects are more rare and tend to be smaller in size.
Type III: A large defect combining Types I and II. Consists of extension of the defect into the right pulmonary artery with anomalous origin of the right pulmonary artery from the ascending aorta (secondary to unequal septation of the aorto-pulmonary trunk during development) with a well-formed inferior rim but little superior rim and involves the majority of the ascending aorta. This type is more frequently linked with other cardiac anomalies.
Type IV: This is an intermediate defect which has adequate superior and inferior rims and are most suitable for possible device closure.
Goals of Echocardiography Exam
Echocardiography is the gold-standard diagnostic tool for aorto-pulmonary window. As patients with AP window are likely to present earlier in life due to symptomatic CHF from significant left-to-right shunting or cyanosis due to the higher risk of associated CHD lesions, a high-frequency probe is recommended in these neonates for evaluation.
Important views for transthoracic echocardiography include:
- Parasternal short-axis 2D and color Doppler: This view allows visualization of both MPA and aorta to evaluate for communications between these structures. Color Doppler is important to confirm that a communication truly exists and there is not just artifactual drop-out in great arterial vessel walls. This view can also help to identify extent of involvement of the RPA. Size measurements of the AP window defect can be made in this view.
- Subcostal short-axis and long-axis views: It is important to evaluate the great arteries and AP window defect in the subcostal plane. These views not only allow for evaluation of the location of the AP window defect, but also: distance from both semilunar valves, extent/presence of the inferior and superior rims, extent of involvement of the ascending aorta, and if involvement of the right pulmonary artery exists, all of which will affect surgical management. In this view, the great arteries association, ventricular septum, pulmonary valve morphology, and right ventricular outflow tract can also be evaluated since AP window can be seen with associated cardiac lesions (ie - VSD, TOF, TGA).
- Suprasternal notch views: 2D and color sweeps from this view between the aortic arch and main pulmonary artery not only allow for evaluation of shunting across the AP window between the great arteries, but allows for thorough evaluation of the arch due to incidence of association of AP window with arch anomalies (type A interrupted aortic arch or coarctation).